 Spanish Version
Spanish Version
|
|
|
|
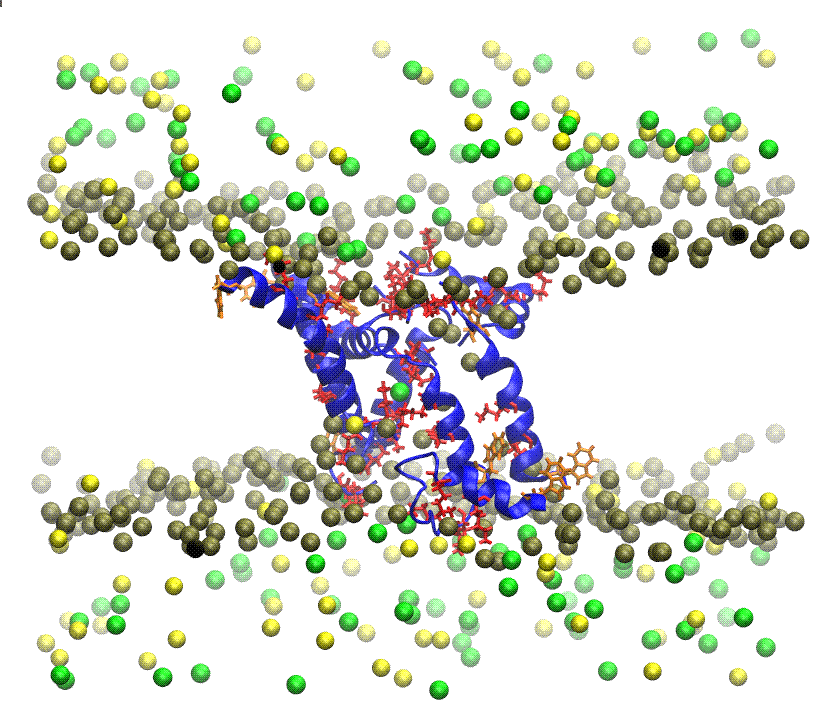
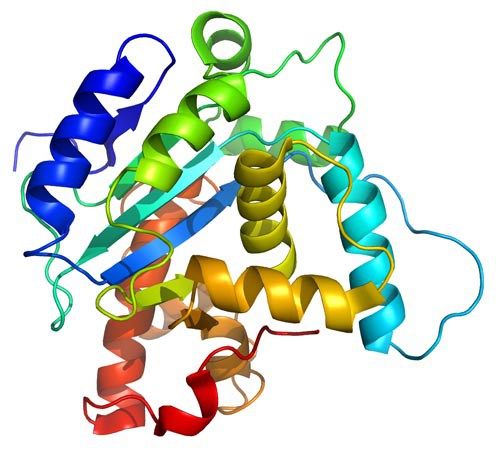
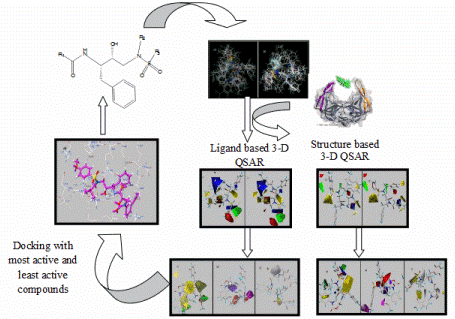
MEMBRANE PROTEINS There are two types: (1) Integral proteins: are those that cross the membrane and appear on both sides of the phospholipid bilayer. Most of these proteins are glycoproteins, proteins that have several monosaccharides attached. The carbohydrate part of the molecule is always facing to the cell exterior, and (2) Peripheral proteins: these are not extended across the width of the bilayer but are attached to its inner or outer surfaces and are easily separated from it. The nature of Membrane Proteins determines its function: Channels: whole proteins (usually glycoproteins) which act as pores by which certain substances can enter or leave the cell, Carriers: are proteins that change shape to give way to certain products, Receptors: are integral proteins that recognize certain molecules to which they are attached or fixed. These proteins can identify a hormone, a neurotransmitter or a nutrient that is important for the cell function. The molecule that binds to the receptor is called ligand, Enzymes: can be integral or peripheral and serve to catalyze reactions at the surface of the membrane, Cytoskeleton anchors: are peripheral proteins found in the part of the membrane cytosol which serve to fix the cytoskeleton filaments, Cell identity markers: are glycoproteins and glycolipids characteristic of each individual and which allow the identification of cells from another organism. For example, blood cells have ABO markers that make possible that in blood transfusion only blood of the same type can be compatible. Being in the exterior, the carbohydrate chains of glycoproteins and glycolipids form a kind of cover called glicocalix.  ANTIMICROBIAL PEPTIDES Resistance to multidrug antibiotics is an increasing severe worldwide public health problem according to reports from the World Health Organization. Thus, there is a significant and urgent need for the development of new classes of antibiotics that do not induce resistance. To develop such antimicrobial compounds, we must look towards agents with new mechanisms of action. The membrane permeabilizing antimicrobial peptides (AMPs) are good candidates because they act without high specificity towards a given protein target, which in turn reduces the probability of induced resistance. Understanding the permeabilization mechanism of the membrane is crucial for the development of AMPs as useful antimicrobial agents. PAMs are a class of evolutionarily conserved molecules with antibiotic properties which occur naturally and are found as the first line of defense of all multicellular organisms. Many PAMs have been isolated from a wide variety of sources. These proteins have shown a wide range of activities to directly kill bacteria, yeasts, fungi, viruses and even cancer cells. Insects and plants deploy AMPs primarily as antibiotics to protect themselves from potentially pathogenic organisms, even though microbes also produce PAMs to defend their niches. In eukaryotes organisms, AMPs are also referred to as "defense peptides", making emphasis on their additional immunomodulatory activities. Since the AMPs have structures and notably different activity profiles, the mechanisms by which the AMPs exert their effect are not well studied at the molecular level. Most PAMs are typically made from 10 to 50 amino acid residues, and are cationic molecules that often contain a distribution of basic and hydrophobic amino acid residues that are aligned in three dimensions on opposite faces, forming unique structures that are soluble in water. These chemical characteristics are important for understanding the mechanism of microbicidal action. It has been proposed that most of these peptides interact directly with the bacterial membrane forming pores and/or structurally disturbing it causing cell lysis. Folded PAMs can be classified into groups based on their secondary structure: α-helix; β-sheet; and extended AMPs. The α-helical amphipatic AMPs include frog magainin and the extensively studied LL37 peptide of human catelicidin. These peptides exhibit very little secondary structure in aqueous solution, but they adopt an α-helical structure when they enter a non-polar environment, such as that of the bacterial membrane. In order to understand AMPs molecular mechanism and specificity towards its cellular target, it is essential to consider the architecture of cellular envelopes. For example, before AMPs can interact with the cytoplasmic membrane of Gram-positive bacteria, they have to traverse the cell wall composed of wall lipoteichoic acids and peptidoglycans. While the interaction of AMPs with peptidoglycan can facilitate penetration, interaction with anionic teicoic acids can act either as a trap for AMPs or as a ladder for a path to the cytoplasmic membrane. Interaction with cytoplasmic membrane often leads to the segregation of lipids that affect the organization of the membrane domain, and therefore affects the permeability of the membrane, inhibits the processes of cell division or leads to the delocalization of the essential proteins of the peripheral membrane. The ability of AMPs to kill bacteria has generally been associated with their ability to interact with bacterial membranes or cell walls. Given that the AMPs exhibit net positive charge and a high proportion of hydrophobic amino acids, it is assumed that this allows them to selectively bind to negatively charged bacterial membranes. The binding of AMPs to the bacterial membrane generally leads to non-enzymatic rupture. The selectivity of the AMPs towards the different species is due to differences in membrane composition of different microbes and cell types. It has been proposed that AMPs disrupt the order of the membrane bilayer by different processes. All these proposals require, either explicitly or implicitly, that a threshold on AMPs concentration at the the membrane surface before the rupture can occur. The most accepted processes are: (A) Barrel pore. The peptides are inserted perpendicularly into the bilayer, and then they oligomerize to form a pore. The peptides are aligned in the pore lumen in a direction parallel to the phospholipid chains, which remain perpendicular to the bilayer plane. (B) Carpet Mechanism. The peptides are adsorbed in parallel to the bilayer and after reaching sufficient coverage, they produce an effect similar to a detergent which disintegrates the membrane. No specific peptide-peptide interactions are required. (C) Toroidal pore. Similar to barrel pores, peptides are inserted perpendicularly to the bilayer, but instead of packing in parallel to the phospholipid chains, they induce a Local curvature of the membrane in a manner such that the pore lumen is partly covered by the peptides and in part by the head groups of phospholipids. A continuity is established between the interior and exterior layers. (D) Disordered Toroidal Pore. A recent modification to the toroidal pore proposes that less rigid conformations and orientations of the peptides are formed; the lumen of the pores is coated by the head groups of phospholipids. We are interested in studying the mechanism by which isolated AMPs from the Panidinus Imperatorscorpion venom, Pandinin 2 (Pin2) and its synthetic analogue Pin2GVG, are able to form pores in the membranes of Gram-positive and Gram-negative bacteria These studies are being carried out using Molecular Dynamics simulations in the presence of bilayer membrane models, with water as solvent and counterions to simulate the cell environment. IONIC CHANNELS Ionic channels are heteromultimeric complexes formed by the assembly of several proteins, that are totally or partially embedded in the membrane, which we call subunits. When the ion channel is opened, an aqueous pore is formed which extends through the thickness of the membrane. The flow of ions through a channel is passive due to differences in electrical potential or in the ion concentrations, that is, it does not need energy expenditure by the cell. Depending on the type of stimulus for its opening or closure ionic channels can be classified in: (1) voltage-activated channels, (2) ligand-activated channels and (3) mechanosensitive channels. Other types of ion channels are present in all plasma membranes of cells, such as the Sodium and Potassium Pump, whose goal is to eliminate sodium from the cell and to introduce potassium into the cytoplasm. That exchange maintains, through the membrane, the different concentrations between both cations. In this way an intracellular negative electrical potential is generated. This mechanism is concentration gradient thanks to the enzyme ATPase, which acts on ATP in order to obtain the energy necessary for nutrients to traverse the cell membrane and reach the cytoplasm. Voltage-controlled ion channels have three main conformational states: closed, open and inactivated. The forward/backward transitions between these states are called activation/deactivation (between open and closed, respectively), inactivation/reactivation (between inactivated and open, respectively) and recovery of inactivation/inactivation, respectively). Closed and inactivated states are impermeable to ions. Over some 400 million years of evolution, sporpions have perfected peptide toxins that are specifically directed and powerful to families of ionic channels found in mammals and insects. Neurotoxic toxins affect neuromuscular junctions and are able to block with great specificity of the major ion channels (sodium, potassium, chlorine and calcium) of the cells. The venom of Centruroides scorpions consists of low mmolecular weight proteins of ca. 700 DA (polypeptides) which are referred to as scorpamines. The scorpamines quickly reach the general circulation and if they come from very poisonous species, within minutes can kill small mammals. Some of these proteins act at the level of ionic channels delaying the release of Na+ and thus prolonging the action potential, with an increase on the time to integrate the current generated by the excitable membranes of the effector cells, and others increase the release of acetylcholine and catecholamines at the synapses, in both the musculo-skeletal and ganglion neurons Scorpamines are divided in three large families: (1) Short-chain (31 to 39 amino acids) that are specific for blocking potassium channels in different cells of excitable membranes. (2) Those of medium size composed of a large variety of polypeptides (61 to 70 amino acids), whose function is to interfere with the flow of sodium ions through the membranes, this family is the most important since from the medical point of view it is divided into toxins α type that interfere with the closure of the sodium channel and β type that prevent the normal function of the sodium channel opening mechanism (these peptides are the most abundant and are present in the venom of all species of scorpions). (3) Long chain polypeptides that act on the calcium permeability in the excitable membranes and in the intracellular behavior (its structure is less studied but it is known that these peptides are made of more than 130 amino acids). The α-toxins are found in the venoms of scorpions from the Old World (Europe, Africa, Asia), while β-toxins are found in the venoms of the New World scorpions (North and South America). The scorpion venom toxins increase the depolarization of the membrane and the release of neurotransmitters by affecting the activation or inactivation of the major ion channels. studies, differences in binding sites, and ion flow have shown that the scorpion venom toxins active against Na+ channels of vertebrates are of two types: (1) α-toxins which slow down channel inactivation, prolonging the action potential; and (2) β-toxins that move dependent voltage activation to more negative membrane potentials leading to repetitive firing in both muscles and nerves. In both cases, the toxins exert their effect from the extracellular side of the membrane increasing the Na+ entry into the cells. The rapid depolarization phase in the action potential of nerve cells, muscle and heart is due to voltage dependent Na+ channels (Nav). While potassium channels (Kv) are involved in the regulation of various cellular functions whose malfunctioning can lead to diseases in different organs. The family of voltage dependent sodium channels has 9 known members and is numbered from Nav1.1 to Nav1.9. The Na+ voltage dependent channels are composed of an α subunit and one or more β subunits. The α subunit is an integral heteromultimeric protein complex which consists of four homologous domains (D1-D4), each of which contains six α-trans-membrane helical segments (S1-S6, made of 19 to 27 amino acids). The C-and N-terminal ends and the loops between the domains are intracytoplasmic. The segments S5-S6 and the P-loop of each domain form the pore of the channel that crosses the entire membrane and contains the selectivity filter. The K+ channels can be classified according to their topology, that is, taking into account the number of pores and transmembrane (TM) segments present in the channel (termed S in channels Kv and M in non-voltage-activated channels), as well as by taking into account the differences in their functional and pharmacological properties. The K+ channels are made up of the α and β subunit assembly, which forms the pore of the channel, and various β subunits. More than 80 genes encoding various human K+ channels have been identified and cloned . More information can be found consulting the following excellent books: Ion Channels and Their Inhibitors, Voltage-Sensitive Ion Channels, and Voltage Gated Sodium Channels. We are interested in studying the mode of action of CssIV, a toxin isolated from the venom of the Centuroides suffusus suffusus scorpion which affects the brain Nav1.2 channels. Likewise, we are interested in studying the mode of action of discrepine, a toxin isolated from the venom of the Tityus discrepans scorpion which affects potassium channels, preferably blocking K-type A-type current generators in granular cells of cerebellum. These studies are being carried out by homology and molecular modeling methods. PROTEIN FOLDING We are interested in helping to solve the Protein Folding Problem (PFP) which refers to knowing how a protein folds to a 3D structure eccepted as the Native Structure. According to the Anfinsen principle, all the information required to specify the structure of a protein is encoded in the sequence of its amino acids which determines if it is biologically active or not. The mathematical prediction of the 3D structure of proteins is almost impossible, since the number of possible structures that a flexible molecule of protein can adopt is astronomical, although in nature proteins fold in a matter of milliseconds to minutes. Experimental methods for determining the 3D structure of any protein, such as X-ray crystallography and NMR spectroscopy are generally expensive and very time-consuming. So the prediction of the structure of proteins by other means becomes necessary, and this goal has become one of the most important research topics of modern structural biology. During the last thirty years several research groups have made importanat advances to understand how proteins fold in nature. In the last decades, scientists suspect that the response to PFP is in the concept of the proetein free energy. It is considered that the native state of a protein folded in its native structure corresponds to the state of lowest free energy, or to one close to the lower. The energy of a given conformation can be modeled using energy functions which model the interactions between amino acid residues. Thus, the PFP can be reduced to an optimization problem, where the energy function has to be minimized as function of the atomic coordinates. Any mathematical model that describes this process is considered a NP complete problem. Our research group tries to help in solving this problem by applying heuristic methods for the localization of the global minimum of conformational energy of small peptides, whose 3D structure is known, with the purpose of subsequently applying these methods to the structure prediction of larger proteins. In the past we have used heuristics such as Tabu Search, Simulated Annealing, and Genetic Algorithms. We are currently interested in the application of bio-inspired algorithms for the prediction of the tertiary structure of proteins. Bio-inspired algorithms are methods of optimization based on some evolutive metaphor or by simulation of social behavior of insects or humans, these have been efficient search methods or as optimization techniques because they are very effective in solving difficult problems. Among them we have the Ant Colony and the Particle Swarm optimization. In protein structure prediction a statistical potential, or knowledge based potential, is an energetic function derived from an analysis of 3D structures of known proteins reported in the Protein Data Bank (PDB). There are many methods to obtain such potential; two notable methods are the quasi-chemical approximation (due to Miyazawa and Jernigan) and the mean force potential (due to Sippl). The simplest way to represent a mean force potential for the pair interaction of amino acids is a simple consequence of the Boltzmann distribution given by:
P(r) =
1
Z
e-
F(r)
kT
where r is the distance, k is the Boltzmann constant, T is the temperature and Z is the partition function defined as:
Z =
∫
e-
F(r)
kT
dr
The quantity F(r) is the free energy associated to a pairwise system. A simple rearrangement provides the inverse Boltzmann formula which expresses the free energy F(r) as function of P(r):
F(r) = -kT ln P(r) - kT ln Z
To construct a mean force potential, the so-called reference state must be entered with a corresponding distribution QR and a partition function ZR, and calculate the following free energy difference:
ΔF(r) = -kT ln
P(r)
QR(r)
- kT ln
Z
ZR
Typically the reference state results from a hypothetical system in which the interactions between specific amino acids are absent. The second term involving Z and to Z(r) is ignored sisnce it is a constant. In practice P(r) is calculated from the information found in the PDB, while QR(r) is typically obtained from calculations or simulations. For example, P(r) could be the conditional probability of finding the Cβ atoms of alanine and glutamine separated by some distance r, giving rise to a difference of energy ΔF. The total free energy difference of a protein, ΔFT, could be considered as the sum of all pairwise interactions. It is ckear that a low value of ΔFT indicates that the set of proper distances come more likely from a folded structure of the protein than from the reference state. Therefore, these potentials can be used to distinguish between structural models obtained by homology modeling or protein threading. ENANTIODIFFERENCIATION We are also interested in knowing the molecular basis of the process of enantiodifferentiation, process by which protein receptors are able to differentiate between chiral drugs. Our interest in this subject emerged when we learned that between 1959 and 1964 the drug Thalidomide (a chiral molecule) was prescribed to prevent anxiety, insomnia and nausea in pregnant women. This drug caused terrible birth defects. Only in Europe more than 10,000 severely deformed children were born, many of them without legs or arms, because their mothers had taken the drug at the beginning of the pregnancy. It was later discovered that the R isomer had the teragenic effect, while The S isomer had the desired sedative effect. Many pharmaceutical drugs on the market are chiral. To understand the molecular basis of the enantiodifferentiation process our research group applies the technique of molecular dynamics on inclusion complexes of chiral model systems, such as cyclodextrins. QSAR Many of the drugs available today were characterized, in their day, by means of conventional screening techniques in the pharmaceutical industry, which consisted of evaluating a battery a biological tests, as many substances as possible, both of natural origin as synthetic ones. This procedure allows the identification of new "head groups", or prototype molecules belonging to a given structural class and with potential in a specific therapeutic area. Subsequent chemical modifications tend to produce "analogues" of those structures with greater activity or lower incidence of side effects. This method of discovering new agents with active is interesting from the point of view that can transform new classes of compounds in potential drugs but, fundamentally based on trial and error techniques, it is time-consuming and requires many economical resources. The first attempts to increase the probability of synthesizing active analogues or to find a new head group were based on finding correlations between the chemical structure of a series of compounds and their biological activity. From there arose the famous acronyms QSAR, for Quantitative Structure-Activity Relationships, which is now a word of current use in the process of of designing new drugs and in the rationalization of the pharmacological properties of a number of substances. The quantitative structure-activity relationship(or QSAR) is the method by which the chemical structure is quantitatively correlated with a well defined process, such as biological activity (binding of a drug to a receptor) or chemical reactivity(affinity of one substance for another to produce a reaction). When physicochemical properties or structures are expressed by numbers, one can construct a mathematical relationship, or quantitative structure-activity relationship, between the two. The mathematical expression can then be used to predict the response of other chemical structures. The differentiation between drug-like molecules from those similar to drugs is essential for reducing the cost associated with failed drug development. For the development of new innovative molecules with pharmacological potential several in silico approaches have demonstrated its potential for the screening of chemical databases against the desired biological targets. The development of a QSAR model should be considered in terms of: (1) the fundamental chemistry of the set of analogues, including any atypical value (outlier); (2) quantitatively correlate and summarize the relationships between the alterations of the chemical structure and the changes in the ultimate biological objective, to determine the chemical properties that are the most likely determinants of the biological activities of the candidate drug; (3) Optimization of existing clues to improve their biological activities; and (4) prediction of the biological activities of the compounds not yet tested. The most general mathematical form of QSAR is: Activity = f (physicochemical properties and/or structural properties) In QSAR modeling, the numerical representations of the physicochemical and structures of biologically active compounds are used. These representations are called descriptors and are fundamental to discover new molecules with pharmacological potential. The mathematical representation of these descriptors has to be invariant to the size of the molecule and the number of atoms it contains to allow the construction of models using statistical methods. The information coded by the descriptors usually depends on the type of the molecular representation and the algorithm defined for its calculation. Generally the descriptors are classified in: (a) topological indexes, (b) geometric descriptors, (c) constitutional descriptors, (d) thermodynamic descriptors and (e) electronic descriptors. The different approaches to QSAR that have been developed in recent decades allow the calculation of reliable relationships between the variations in the values of the calculated descriptors and the biological activity of a number of chemical molecules; in such a way that they can be used to predict the activity of compounds not yet tested or newly synthesized. The chemical structures used in construction of QSAR models are encoded by a substantial number of molecular descriptors. The model is constructed using only a few descriptors that are valid for compounds closely related. To validate the QSAR models, four strategies are usually adopted: (1) internal validation or cross validation (cross validation) (2) validation by dividing the data set into test (samples) and training compounds; (3) true external validation by application of the model on external data and (4) data scrambling or Y-scrambling. As the use of (Q)SAR models for chemical risk management steadily increases and it is also used for regulatory purposes, it is of crucial importance that QSAR would be able to assess the reliability of predictions. For a given chemical training set the extended chemical descriptor space is called applicability domain. The applicability domain of a QSAR model is the physical-chemical, structural or biological, knowledge or information on which the training set of the model has been developed and for which it is applicable to make predictions for new compounds. Since the QSAR concept was first introduced in 1964, a wide range of methodologies have been invented. Traditional 2D-QSAR methods such as the Free-Wilson and Hansch-Fujita approaches use 2D molecular substituents or fragments and their physicochemical properties to make quantitative predictions. Since then, QSAR has experienced a rapid development and the first novel 3D-QSAR method called molecular field comparative analysis (CoMFA) was introduced by Cramer et al. in 1988. The CoMFA method laid the foundations for the development of other 3D-QSAR methods such as CoMSIA, SOMFA, CoMMA as well as multidimensional (nD)-QSAR methods such as 4D-QSAR, 5D-QSAR, etc., used to address known 3D-QSAR problems such as subjective molecular alignment and bioactive conformation problems. In recent years, methods based on fragments have attracted some attention because to predict molecular properties and activities based on simple molecular fragments is simple, fast and robust. There is a very good review of these methods in the following link. FREE ENERGY The second law of thermodynamics helps us to determine whether a process will be spontaneous and the use of the changes in Gibbs free energy to predict if a reaction will be spontaneous in one or in other sense (or if it is in equilibrium). Free energy constitutes the most important thermodynamic quantity to understand how chemical species are recognized, associated or reacted. Examples of problems that required knowledge of underlying free energy behavior include conformational equilibria and molecular association, partition between immiscible liquids, receptor-drug interaction, association protein-protein, protein-DNA and stability of proteins. The receptors are molecules of a protein nature, usually associated with cell membranes. These proteins may be associated with other elements that determine the effect. The receptor is responsible for recognizing the ligand, has the ability to recognize it selectively and once the union is realized the effect is triggered, which is mediated by other structures attached to the receptor. A ligand is any molecule that specifically binds to a receptor whose characteristics are: (A) Variable chemical size and nature (B) The binding site is specific (C) The union is reversible -weak Interactions- the equilibrium is reached between: Protein-Ligand ⇆ Proteinfree + Ligandfree
At the end of the 19th century it was proposed (Emil Fischer) that the union is of the type "lock and key", that is to say, that the forms of the receptor and ligand binding site are strictly complementary. Later, in 1958 this concept was redefined (Daniel Koshland) proposing that in the initial phase of the union the complementarity is relatively poor, but after the initial binding the protein undergoes a conformational change which "adjusts" the shape of the binding site to that of the ligand. This is the hypothesis of the "induced fit". Once the ligand has been attached to its receptor one of the following actions is carried out: (1) The ligand is unchanged (transport). (2) The ligand is chemically modified (enzyme) (3) The protein undergoes a conformational change. For the case where the protein has a single binding site, and if initially the protein (P) and the ligand (L) are free in the medium, after a time will have established an equilibrium between P and L and the protein-ligand complex ligand, the chemical balance can be written as follows:
P + L
⇄K1
K-1
PL
Where K1 is the association kinetic constant whose dimensions are (mole-1 s-1), and K -1 is the rate constant of the dissociation reaction of PL, whose dimension is the inverse of time (s-1). Thus: V-1 = V1
K -1[PL] = K1[P][L]
K -1
K1
=
[P][L]
[PL]
= Kd
Kd is the so-called Dissociation Constant and corresponds to the equilibrium constant of the PL complex dissociation. Do not confuse with K -1 which is the velocity constant for the dissociation reaction. Kd has units of concentration.
Kd =
[P]free[L]
[PL]
Therefore, the value of Kd for specific protein and ligand depends critically on the experimental conditions such as: pH, ionic strength, temperature or presence of other solutes in the media. This is logical if we remember that all the factors mentioned above influence the 3D structure of proteins, and that the binding site is a specific area with a defined structure within the protein molecule. Relationship between Kd and the free energy variation in the association. If we need to know the free energy variation that occurs in the formation of the [PL] complex, one has to take into account the initial and final conditions in the protein and ligand system (without forgetting the molecules of water in which both are dissolved), since the system can be in two states: (I) the protein and the ligand are separated and interact with the water molecules, and (II) the PL complex and the water molecules that were previously bound to the protein and the ligand are now part of the bulk of the solvent. It is very important to consider that the variation of the free energy depends not only on the ionic bonds, the hydrogen bonds and the van der Walls forces formed between the protein and the ligand, but also of the interactions both have with the water molecules and those between the water molecules. The variation of free energy under standard conditions (concentration of 1 M reactants, pH 7.0, 25 C) is related to the equilibrium constant of the reaction by the expression: ΔG0' = -RT lnKeq; being R the gas constant ant T the absolute temperature. Therefore, if we want to know the energy that is released when the protein and the ligand are associated, assuming that the system is in equilibrium, and taking into account that usually we are going to employ Kd, which is the dissociation equilibrium constant, we would have to employ the following relation: ΔG0' = -RT lnKassociation =
+RT lnKd;
For a better understanding of the theory and applications of the calculation of Free Energy in Chemistry and Biology is recommended to review the contents of the book Free Energy Calculations: Theory and Applications in Chemistry and Biology. |
|
Copyright © Ramón Garduño-Juárez. All Rights Reserved. |
 E-mail: | Mayo 2017
E-mail: | Mayo 2017


 Home
Home