 English Version
English Version
|
|
|
|
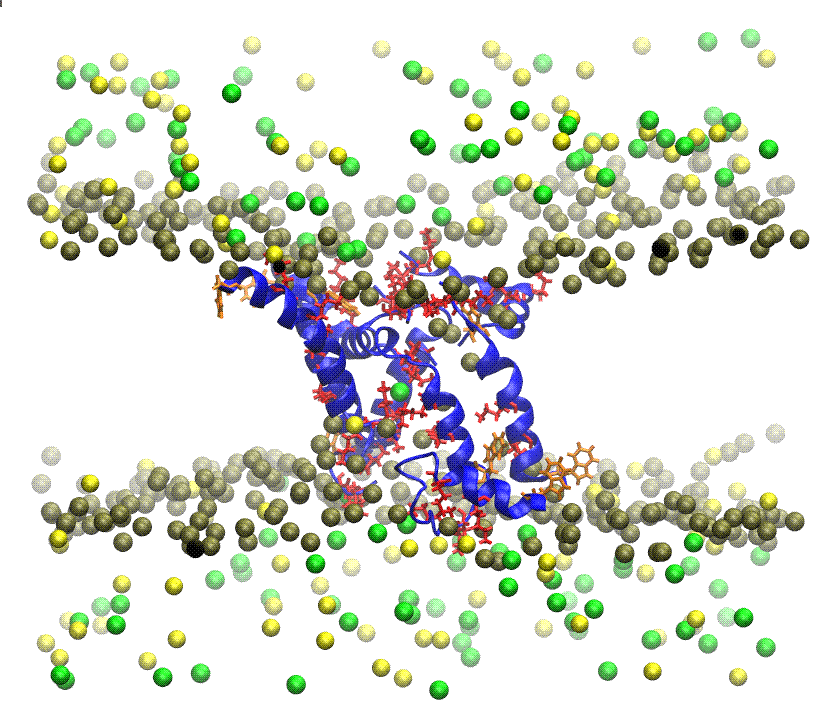
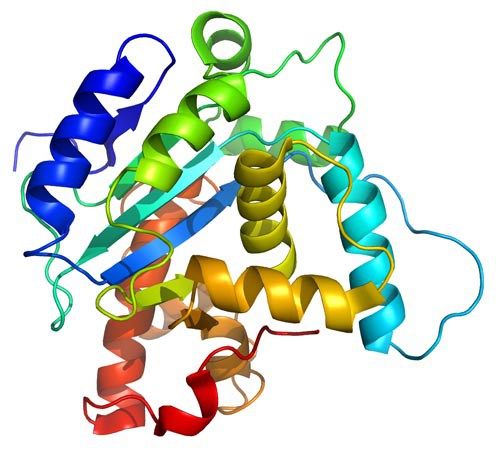
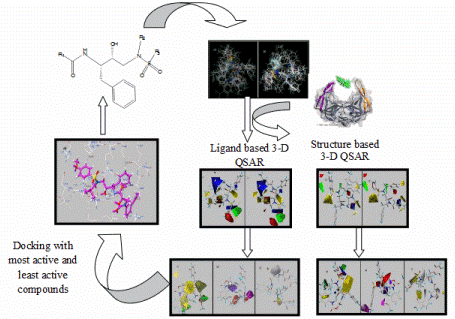
PROTEÍNAS DE MEMBRANA Las hay de dos tipos: (1) Proteínas integrales: son aquellas que cruzan la membrana y aparecen a ambos lados de la capa de fosfolípidos. La mayor parte de estas proteínas son glicoproteinas, proteínas que tienen unidos uno varios monosacáridos. La parte de carbohidrato de la molécula está siempre de cara al exterior de la célula, y (2) Proteínas periféricas: estás no se extienden a lo ancho de la bicapa sino que están unidas a las superficies interna o externa de la misma y se separan fícilmente de la misma. La naturaleza de las proteínas de membrana determina su función: Canales: proteínas integrales (generalmente glicoproteínas) que actúan como poros por los que determinadas sustancias pueden entrar o salir de la célula, Transportadoras: son proteínas que cambian de forma para dar paso a determinados productos, Receptores: son proteínas integrales que reconocen determinadas moléculas a las que se unen o fijan. Estas proteínas pueden identificar una hormona, un neurotransmisor o un nutriente que sea importante para la función celular. La molécula que se une al receptor se llama ligando, Enzimas: pueden ser integrales o periféricas y sirven para catalizar reacciones a en la superficie de la membrana, Anclajes del citoesqueleto: son proteínas periféricas que se encuentran en la parte del citosol de la membrana y que sirven para fijar los filamentos del citoesqueleto, Marcadores de la identidad de la célula: son glicoproteínas y glicolípidos característicos de cada individuo y que permiten identificar las células provenientes de otro organismo. Por ejemplo, las células sanguíneas tienen unos marcadores ABO que hacen que en una transfusión sólo sean compatibles sangres del mismo tipo. Al estar hacia el exterior las cadenas de carbohidratos de glicoproteínas y glicolípidos forman una especie de cubierta denominada glicocalix.  PÉPTIDOS ANTIMICROBIANOS La resistencia a los antibióticos multidrogas es un problema de salud pública cada vez más grave en todo el mundo según reportes de la Organización Mundial de la Salud. Así, existe una necesidad significativa y urgente para el desarrollo de nuevas clases de antibióticos que no inducen resistencia. Para desarrollar tales compuestos antimicrobianos, debemos mirar hacia agentes con nuevos mecanismos de acción. Los péptidos antimicrobianos permeabilizantes de membrana (PAMs) son buenos candidatos porque actúan sin alta especificidad hacia un objetivo de proteína, lo que reduce la probabilidad de resistencia inducida. Comprender el mecanismo de permeabilización de la membrana es crucial para el desarrollo de PAMs en agentes antimicrobianos útiles. Los PAMs son una clase de moléculas conservadas evolutivamente y con propiedades antibióticas que ocurren naturalmente y que se encuentran como la primera línea de defensa de todos organismos multicelulares. Se han aislado una gran cantidad de PAMs de una gran variedad de fuentes. Estas proteínas han mostrado una amplia gama de actividades para matar directamente bacterias, levaduras, hongos, virus e incluso a células cancerosas. Los insectos y las plantas despliegan a los PAMs principalmente como un antibiótico para protegerse de organismos potencialmente patógenos, a pesar de que los microbios también producen PAMs para defender sus nichos. En organismos eucariotas superiores, los PAMs también son referidos como "péptidos de defensa", haciendo hincapié en sus actividades inmunomoduladoras adicionales. Dado que los PAMs tienen estructuras y perfiles de actividad notablemente diferentes, los mecanismos por los cuales los PAMs ejercen su efecto no están bien estudiados a nivel molecular. La mayoría de los PAMs típicamente están hechos de entre 10 a 50 residuos de aminoácido, y son moléculas catiónicas que con frecuencia contienen una distribución de aminoácidos básicos y residuos hidrofóbicos que se alinean en tres dimensiones en caras opuestas, formando estructuras únicas que son solubles en agua. Estas características químicas son importantes para comprender el mecanismo de acción microbicida. Se ha propuesto que la mayoría de estos péptidos interactuan directamente con la membrana de la bacteria formando poros y/o perturbando estructuralmente a ésta ocasionando la lisis celular. Los PAMs plegados pueden clasificarse en grupos basados en su estructura secundaria: α-helicoidal; hoja-β; y PAMs extendidos. Los PAMs anfipáicos de α-hélice incluyen a la magainina de la rana y al extensamente estudiado péptido LL37 de la catelicidina humana. Estos péptidos exhiben muy poca estructura secundaria en disolución acuosa, pero adoptan una arquitectura α-helicoidal cuando entran en un entorno no polar, como el de la membrana bacteriana. Con el fin de comprender su mecanismo molecular y especificidad de su objetivo celular, es esencial considerar la arquitectura de las envolturas celulares. Por ejemplo, antes de que los PAMs puedan interactuar con la membrana citoplasmática de las bacterias Gram-positivas, éstos tienen que atravesar la pared celular compuesta por los ácidos lipoteicoicos de pared y los peptidoglicanos. Si bien la interacción de los PAMs con el peptidoglicano puede facilitar la penetración, la interacción con los ácidos teicoicos aniónicos puede actuar ya sea como una trampa para los PAMs o como una escalera para una ruta a la membrana citoplasmática. La interacción con la membrana citoplasmática con frecuencia conduce a la segregación de los lípidos que afecta la organización del dominio de la membrana, y por lo tanto afecta la permeabilidad de la membrana, inhibe los procesos de división celular o conduce a la deslocalización de las proteínas esenciales de la membrana periférica. La capacidad de los PAMs para matar bacterias generalmente se ha asociado a su capacidad para interactuar con membranas bacterianas o paredes celulares. Dado que los PAMs exhiben carga positiva neta y una alta proporción de aminoácidos hidrofóbicos, se supone que esto les permite unirse selectivamente a las membranas bacterianas cargadas negativamente. La unión de los PAMs a la membrana bacteriana generalmente conduce a la ruptura no enzimática. La selectividad de los PAMs hacia las diferentes especies se debe a las diferencias en la composición de la membrana de los diferentes microbios y tipos de células. Se ha propuesto que los PAMs interrumpen el orden de la bicapa de la membrana por diferentes procesos. Todas estas propuestas requieren, ya sea explícita o implícitamente, que se cruze una concentración umbral de éstos en la membrana para que la ruptura pueda ocurrir. Los procesos más aceptados son: (A) Poro de Barril. Los péptidos se insertan perpendicularmente en la bicapa, se asocian y forman un poro. Los péptidos se alinean en el lumen del poro en una dirección paralela con respecto a las cadenas de fosfolípidos, que permanecen perpendiculares al plano de bicapa. (B) Mecanismo de Alfombra. Los péptidos se adsorben de forma paralela a la bicapa y, después de alcanzar una cobertura suficiente, producen un efecto parecido a un detergente que desintegra a la membrana. No se requieren interacciones específicas de péptido-péptido. (C) Poro Toroidal. De forma parecida a los poros de barril, los péptidos se insertan perpendicularmente en la bicapa, pero en lugar de empacarse en paralelo a las cadenas de fosfolípidos, éstos inducen una curvatura local de la membrana de una manera tal que el lumen del poro es cubierto en parte por los péptidos y en parte por los grupos principales de los fosfolípidos. Se establece una continuidad entre las capas interior y exterior. (D) Poro Toroidal Desordenado. Una reciente modificación al poro toroidal propone que se forman conformaciones y orientaciones menos rígidas de los péptidos; el lumen de los poros está revestido por los grupos principales de los fosfolípidos. Estamos interesados en estudiar el mecanismo por el cual el PAM aislado del veneno del alacrán Panidinus imperator, Pandinina 2 (Pin2) y su analogo sintetico Pin2GVG, son capaces de formar poros en las membranas de bacterias Gram-positivas y Gram-negativas. Estos estudios los estamos realizando mediante simulaciones de Dinámica Molecular en la presencia de modelos de membrana bicapa, con agua como disolvente y de contraiones para simular el ambiente que rodea a las células. CANALES IÓNICOS Los canales iónicos son complejos heteromultiméricos formados por el ensamblaje de varias proteínas que se encuentran embebidas total o parcialmente en la membrana a las que denominamos subunidades . Cuando el canal iónico se abre, se forma un poro acuoso que se extiende a través del espesor de la membrana. El flujo de iones a través de un canal es pasivo debido a diferencias en el potencial eléctrico o en las concentraciones, es decir, no necesita de gasto metabólico energético por parte de la célula. Los canales iónicos se pueden clasificar en función del tipo de estímulo para su abertura o cierre en: (1) canales activados por voltaje, (2) canales activados por ligandos y (3) canales mecanosensibles. Otro tipo de canales ionicos están presente en todas las membranas plasmáticas de las células, como la bomba de sodio y potasio, cuyo objetivo es eliminar sodio de la célula e introducir potasio en el citoplasma. Ese intercambio permite mantener, a través de la membrana, las diferentes concentraciones entre ambos cationes. De esa forma se genera un potencial eléctrico negativo intracelular. Este mecanismo se produce en contra del gradiente de concentración gracias a la enzima ATPasa, que actúa sobre el ATP con el fin de obtener la energía necesaria para que los nutrientes puedan atravesar la membrana celular y llegar al citoplasma. Los canales ionicos controlados por voltaje tienen tres estados conformacionales principales: cerrados, abiertos e inactivados. Las transiciones de avance/retroceso entre estos estados se denominan activación/desactivación (entre abierto y cerrado, respectivamente), inactivación/reactivación (entre inactivado y abierto, respectivamente) y recuperación de inactivación/inactivación de estado cerrado, respectivamente. Los estados cerrados e inactivados son impermeables a los iones. A lo largo de unos 400 millones de años de evolución, los alacránes han perfeccionado toxinas peptidicas dirigidas de forma específica y potente a familias de canales iónicos de mamíferos e insectos. Las toxinas neurotóxicas afectan a las uniones neuromusculares y son capaces de bloquear con gran especificidad los principales canales iónicos (sodio, potasio, cloro y calcio) de las células. El veneno de los alacranes Centruroides está formado por proteínas de bajo peso molecular de ca. 700 DA (polipéptidos) a las que se les conoce como escorpaminas. Las escorpaminas llegan rápidamente a la circulación general y si son provenientes de especies muy venenosas, en minutos pueden matar mamíferos pequeños. Algunas de estas proteínas actúan a nivel de los canales iónicos retardando la liberación de Na+ y prolongando así el potencial de acción, con aumento del tiempo para integrar la corriente generada por las membranas excitables de las células efectoras, y otras incrementan la liberación de acetilcolina y catecolaminas a nivel sináptico, tanto en las terminaciones musculo-esqueléticas como en las neuronas ganglionares. Entre las escorpaminas se encuentras tres grandes familias: (1) Las de cadena corta (31 a 39 aminoácidos) que son específicas para el bloqueo de canales de potasio en diferentes células de membranas excitables. (2) Las de de cadena mediana compuestas por una gran variedad de polipéptidos (61 a 70 aminoácidos), cuya función es interferir con el flujo de iones de sodio a través de las membranas, esta familia es la más importante desde el punto de vista medico y se divide en toxinas tipo α que interfieren con el cierre del canal de sodio y las de tipo β que impiden el funcionamiento normal del mecanismo de apertura de canales de sodio (estos péptidos son los más abundantes y están presenten en el veneno de todas las especies de escorpiones). (3) Las de cadena larga compuestas por polipéptidos que actúan sobre la permeabilidad al calcio en las membranas excitables y en el comportamiento intracelular (su estructura está menos estudiada pero se sabe que contienen mas de 130 aminoácidos). Las α-toxinas se encuentran en los venenos de los alacránes del Viejo Mundo (Europa, Africa, Asia), mientras que las β-toxinas se encuentran en los venenos de los alacránes del Nuevo Mundo (Norte y Sur-America). Las tóxinas del veneno del alacrán incrementan la depolarización de la membrana y la liberación de neurotransmisores al afectar la activación o inactivación de los principales canales iónicos. Estudios electrofisiológicos, diferencias en los sitios de unión, y flujo de iones han demostrado que las tóxinas del veneno de alacrán activas contra los canales de Na+ de los vertebrados son de dos tipos: (1) α-toxinas que alentan la inactivación del canal, prolongando el potencial de acción; y (2) β-toxinas que mueven la activación voltaje dependiente a potenciales de membrana más negativos llevando a disparos repetitivos tanto en músculos como en nervios. En ambos casos, las tóxinas ejercen su efecto desde el lado extracelular de la membrana y la entrada de Na+ hacia las células aumenta. La rapida fase de depolarización del potencial de acción de las células nerviosas, de músculo y de corazón se debe a los canales de Na+ dependientes de voltaje (Nav). Mientras que Los canales de potasio dependientes de voltaje (Kv) están implicados en la regulación de diversas funciones celulares por lo que un mal funcionamiento de éstas puede conducir a enfermedades en distintos órganos. La familia de los canales de sodio dependiente del potencial tiene 9 miembros conocidos y son numerados desde Nav1.1 a Nav1.9. Los canales de Na+ voltaje dependientes están compuestos por una subunidad α y una o más subunidades β. La subunidad α es un complejo heteromultimérico proteico integral que consta de cuatro dominios homólogos (D1-D4), cada uno de los cuales contiene seis segmentos α-hélice transmembranal (S1-S6, de 19 a 27 aminoácidos). Los extremos C-y N-terminal y los lazos de unión de los dominios entre sí son intracitoplasmáticos. Los segmentos S5-S6 y el lazo P de cada dominio forman el poro del canal que penetra en el interior de la membrana y contiene el filtro de selectividad del mismo. Los canales de K+ se pueden clasificar atendiendo a su topología, es decir, al número de poros y de segmentos transmembrana (TM) del canal (denominados S en los canales Kv y M en los canales no activados por voltaje), así como atendiendo a las diferencias en sus propiedades funcionales y farmacológicas. Los canales de K+ están formados por el ensamblaje de subunidades α y β que forman el poro del canal y diversas subunidades β. Se han identificado y clonado más de 80 genes que codifican diversos canales de K+ humanos. Estamos interesados en estudiar la interacción de la toxina CssIV aislada del veneno del alacrán Centuroides suffusus suffusus que afecta a los canales Nav1.2 de cerebro. Asimismo, estamos interesados en estudiar al interacción de la toxina discrepina con canales de potasio. La discrepina es una toxina aislada del veneno del alacrán Tityus discrepans y ha mostrado que puede bloquear de manera preferente canales Kv generadores de corriente tipo-A en células granulares de cerebelo. Estos estudios los estamos realizando mediante simulaciones de Dinámica Molecular en la presencia de modelos de membrana bicapa, con agua como disolvente y de contraiones para simular el ambiente que rodea a las células. PLEGADO DE PROTEÍNAS Estamos interesados en contribuir a resolver el Problema del Plegado de Proteínas (PPP) el cual se refiere a saber como una proteína se pliega a una estructura 3D conocida como la estructura nativa. Según el principio de Anfinsen, toda la información requerida para especificar la estructura de una proteína está codificada en la secuencia de sus amino ácidos, la cual determina si ésta es biológicamente activa o no. La predicción de la estructura tridimensional de proteínas es casi imposible, ya que el número de posibles estructuras que puede adoptar una molécula de proteína es astronómico, sin embargo en la naturaleza las proteínas se pliegan en cuestión de milisegundos a minutos. Los métodos experimentales para determinar la estructura 3D de cualquier proteína, tales como la cristalografía de Rayos-X y la espectroscopía de la RMN, generalmente son caros y muy tardados. Así que la predicción de la estructura de las proteínas por otros medios se hace necesaria, esta tarea se ha convertido en uno de los tópicos de investigación más importantes de la biología estructural moderna. Durante los últimos treinta años varios grupos de investigación han realizado avances importantes para entender como se pliegan las proteínas en la naturaleza. En las últimas décadas, los científicos sospechan que la respuesta al PPP está en el concepto de la energía libre de una proteína. Se considera que el estado nativo de una proteína plegada en su estado nativo corresponde al estado de más baja energía libre, o a uno de los más bajos. La energía de una conformación puede ser modelada empleando funciones de energía que modelan las interacciones entre los residuos de aminoácidos. Así, el PPP se puede reducir a un problema de optimización, donde la función de energía tiene que ser minimizada en función de las coordenadas atómicas. Cualquier modelo matemático que describa este proceso se le considera un problema NP completo. Nuestro grupo de trabajo intenta dar respuesta a esta pregunta al aplicar métodos heuristicos para la localización del mínimo global de energía conformacional de péptidos pequeños, cuya estructura tridimensional es conocida, con el propósito de posteriormente aplicar estos métodos a la predicción de proteínas mas grandes. En el pasado hemos utilizado heurísticas tales somo la Búsqueda Tabu, el Recocido Simulado, y los Algoritmos Genéticos. Actualmente estamos interesados en la aplicación de algoritmos bio-inspirados para la predicción de la estructura terciaria de proteínas. Los algoritmos bio-inspirados son métodos de optimización basados en alguna metáfora evolutiva o por simulación de comportamientos sociales de insectos o humanos, éstos se han vuelto populares para búsquedas eficientes y como técnicas de optimización debido a que son muy efectivos en la solución de problemas difíciles. Entre ellos tenemos a la optimización por Colonia de Hormigas, y por Enjambre de Partículas. En la predicción de la estructura de una proteína un potencial estadístico, o potencial basado en el conocimiento, es una función energética derivada de un análisis de estructuras 3D de proteínas conocidas reportadas en el Protein Data Bank (PDB). Existen muchos métodos para obtener tales potenciales; dos métodos notables son la aproximación cuasi-química (debido a Miyazawa y Jernigan) y el potencial de la fuerza media (debido a Sippl). La manera más simple de representar un potencial de fuerza media para la interacción por pares de amino ácidos es una consecuencia simple de la distribución de Boltzmann dada por:
P(r) =
1
Z
e-
F(r)
kT
donde r es la distancia, k es la constante de Boltzmann, T es la temperatura y Z es la función de partición definida como:
Z =
∫
e-
F(r)
kT
dr
La cantidad F(r) es la energía libre asociada al sistema por pares. Un simple rearreglo da como resultado la fórmula inversa de Boltzmann que expresa la energía libre F(r) en función de P(r):
F(r) = -kT ln P(r) - kT ln Z
Para construir un campo de fuerza media se debe introducir el asi llamado estado de referencia con una distribución correspondiente QR y una función de partición ZR, y calcular la siguiente diferencia de energía libre:
ΔF(r) = -kT ln
P(r)
QR(r)
- kT ln
Z
ZR
Típicamente el estado de referencia resulta de un sistema hipotético en el cual las interacciones específicas entre los amino ácidos están ausentes. El segundo término que involucra a Z y a Z(r) se ignoran ya que es una constante. En la práctica P(r) se calcula de la base de datos del PDB, mientras que QR(r) se obtiene típicamente de cálculos o simulaciones. Por ejemplo, P(r) podría ser la probabilidad condicional de encontrar a los átomos Cβ de alanina y de glutamina separados por cierta distancia r, dando lugar a una diferencia de energía ΔF. El total de la diferencia de energía de una proteína, ΔFT, puede ser considerada como la suma de todas las interacciones por pares. Queda claro que un valor bajo de ΔFT indica que el conjunto de distancias proviene más probablemente de una estructura plegada de la proteína que del estado de referencia. Por lo tanto, estos potenciales se pueden usar para distinguir entre modelos estructurales obtenidos por modelado por homología y por enhebrado. ENANTIODIFERENCIACIÓN También estamos interesados en conocer las bases moleculares del proceso de enantiodiferenciación de drogas quirales. Nuestro interés en este tema surgío cuando nos enteramos que entre 1959 y 1964 se administró la Talidomida (una molécula quiral) para evitar la ansiedad, el insomnio y las náuseas en las mujeres embarazadas. Esta droga causó terribles malformaciones de nacimiento. Solo en Europa nacieron mas de 10,000 niños gravemente deformes, muchos de ellos sin piernas ni brazos, porque sus madres habían tomado la droga al comienzo del embarazo. Posteriormente se supo que el isomero R tiene el efecto teragénico, mientras que el isomero S tiene el efecto sedativo buscado. Muchos medicamentos en el mercado son quirales. Para entender las bases moleculares del proceso de enantiodiferenciación nuestro grupo de investigación aplica la técnica de la dinámica molecular sobre complejos de inclusión de sistemas quirales modelo, como lo son las ciclodextrinas. QSAR Muchos de los fármacos hoy disponibles en la industria farmacéutica fueron caracterizados en su día mediante técnicas de cribado convencionales, consistentes en evaluar en una batería, lo más amplia posible, de ensayos biológicos el mayor número posible de sustancias, tanto de origen natural como sintético. Con este procedimiento se consiguen identificar nuevos "cabezas de serie", o moléculas prototipo pertenecientes a una clase estructural determinada y con potencial en una área terapéutica concreta. Modificaciones químicas subsiguientes tienden a producir "análogos" de esas estructuras con una mayor actividad o una menor incidencia de efectos colaterales. Este método de descubrimiento de nuevos agentes con actividad biológica es interesante desde el punto de vista de que puede convertir a nuevas clases estructurales de compuestos en fármacos potenciales pero, al estar basado fundamentalmente en técnicas de ensayo y error, consume mucho tiempo y requiere grandes recursos económicos. Los primeros intentos dirigidos a incrementar la probabilidad de sintetizar un análogo activo o de encontrar un nuevo cabeza de serie se basaron en encontrar correlaciones entre la estructura química de una serie de compuestos y su actividad biológica. De ahí surgieron las famosas siglas QSAR, acrónimo de Quantitative Structure-Activity Relationships, que es hoy día una palabra de uso corriente tanto en el proceso de diseño de nuevos fármacos como en la racionalización de las propiedades farmacológicas de una serie de sustancias. La relación cuantitativa estructura-actividad (o QSAR) es el método por el cual la estructura química se correlaciona cuantitativamente con un proceso bien definido, como la actividad biológica (unión de un fármaco con un receptor) o la reactividad química (afinidad de una sustancia por otra para que produzcan una reacción). Cuando las propiedades fisicoquímicas o las estructuras se expresan mediante números, se puede construir una relación matemática, o relación cuantitativa estructura-actividad, entre las dos. La expresión matemática puede entonces usarse para predecir la respuesta de otras estructuras químicas. La diferenciación entre moléculas tipo fármacos de aquellas semejantes a fármacos es esencial para reducir el costo asociado con el desarrollo fallido de fármacos. Varios enfoques in silico han demostrado su potencial para el cribado de bases de datos químicos contra las dianas biológicas deseadas para el desarrollo de nuevas moleculas innovadoras con potencial farmacológico. El desarrollo de un modelo QSAR debe considerarse en términos de: (1) la química fundamental del conjunto de análogos, incluyendo cualquier valor atípico; (2) el correlacionar cuantitativamente y resumir las relaciones entre las alteraciones de la estructura química y los cambios relevantes en el objetivo biológico final, para determinar las propiedades químicas que son los determinantes más probables de las actividades biológicas del fármaco candidato; (3) la optimización de las pistas existentes para mejorar sus actividades biológicas; y (4) predicción de las actividades biológicas de los compuestos sin probar. La forma matemática más general de QSAR es: Actividad = f(propiedades fisicoquímicas y/o propiedades estructurales) En el modelado de QSAR se emplean las representaciones numéricas de las propiedades fisicoquímicas y estructurales de compuestos biológicamente activos. Estas representaciones reciben el nombre de descriptores y son fundamentales para descubrir nuevas moléculas con potencial farmacológico. La representación matemática de estos descriptores tiene que ser invariante al tamaño de la molécula y al número de átomos que contiene para permitir la construcción de modelos con métodos estadísticos. La información codificada por los descriptores generalmente depende del tipo de representación molecular y del algoritmo definido para su cálculo. Generalmente los descriptores están clasificados en: (a) índices topológicos, (b) descriptores geométricos, (c) descriptores constitucionales, (d) descriptores termodinámicos y (e) descriptores electrónicos. Los diferentes enfoques de QSAR que se han desarrollado en las últimas décadas permiten determinar relaciones confiables entre las variaciones en los valores de los descriptores calculados y la actividad biológica de una serie de moléculas químicas; de tal manera que pueden usarse para predecir la actividad de compuestos no probados o recién sintetizados. Las estructuras químicas utilizadas en la construcción de modelos QSAR están codificadas por un número sustancial de descriptores moleculares. El modelo se construye utilizando sólo unos pocos descriptores que son válidos para compuestos estrechamente relacionados. Para validar los modelos QSAR se adoptan habitualmente cuatro estrategias: (1) validación interna o validación cruzada (validación cruzada); (2) validación por división del conjunto de datos en compuestos (muestras) de entrenamiento y de prueba; (3) validación externa verdadera por aplicación del modelo sobre datos externos y (4) aleatorización de datos o Y-scrambling. Como el uso de los modelos (Q)SAR para gestión del riesgo químico aumenta de modo constante y también se emplea para propósitos de regulación, es de crucial importancia que el modelo QSAR sea capaz de evaluar la fiabilidad de las predicciones. El espacio de descriptores químicos extendido para un determinado conjunto de entrenamiento de compuestos químicos se llama dominio de aplicabilidad. El dominio de aplicabilidad de un modelo QSAR es el espacio físico-químico, estructural o biológico, conocimiento o información sobre el cual se ha desarrollado el conjunto de entrenamiento del modelo y para el cual es aplicable hacer predicciones para nuevos compuestos. Desde que el concepto QSAR fue introducido por primera vez en 1964, una amplia gama de metodologías se han inventado. Los métodos tradicionales 2D-QSAR tales como los enfoques de Free-Wilson y Hansch-Fujita utilizan sustituyentes o fragmentos moleculares 2D y sus propiedades fisicoquímicas para realizar predicciones cuantitativas. Desde entonces, QSAR ha experimentado un rápido desarrollo y el primer método 3D-QSAR novedoso llamado análisis comparativo de campo molecular (CoMFA) fue introducido por Cramer et al. en 1988. El método CoMFA puso las bases para el desarrollo de otros métodos 3D-QSAR tales como CoMSIA, SOMFA, CoMMA así como métodos multidimensionales (nD)-QSAR tales como 4D-QSAR, 5D-QSAR, etc., usados para abordar problemas conocidos de 3D-QSAR tales como la alineación molecular subjetiva y problemas de conformación bioactiva. En los últimos años, los métodos basados en fragmentos han atraído cierta atención porque el predecir propiedades moleculares y actividades basados en fragmentos moleculares es simple, rápida y robusta. Existe una muy buena revisión de estos métodos en la siguiente liga. ENERGÍA LIBRE La segunda ley de la termodinámica nos ayuda a determinar si un proceso será espontáneo y el uso de los cambios en la energía libre de Gibbs para predecir si una reacción será espontánea en uno o en otro sentido (o si estará en equilibrio). La energía libre constituye la cantidad termodinámica más importante para entender cómo las especies químicas se reconocen, se asocian o reaccionan. Ejemplos de problemas en los que se requiere conocimiento del comportamiento energético libre subyacente incluyen equilibrios conformacionales y asociación molecular, partición entre líquidos inmiscibles, interacción receptor-fármaco, asociación proteína-proteína, proteína-ADN y estabilidad de proteínas. Los receptores son moléculas de naturaleza proteíca, generalmente asociadas a membranas celulares. Estas proteínas pueden estar asociadas a otros elementos que son los que determinan el efecto. El receptor está encargado de reconocer al ligando, tiene la capacidad de reconocerlo selectivamente y una vez que se realiza la unión se desencadena el efecto, el cual está mediado por otras estructuras unidas al receptor. Un ligando es cualquier molécula que se una específicamente a un receptor cuyas características son: (a) Tamaño y naturaleza química variable (b) El sitio de unión es específico (c) La unión es reversible –Interacciones débiles- se alcanza el equilibrio entre: Proteína-Ligando ⇆ Proteínalibre + Ligandolibre
A finales del siglo XIX se propuso (Emil Fischer) que la unión es del tipo "llave y cerradura", es decir, que las formas del sitio de unión del receptor y del ligando son estrictamente complementarias. Posteriormente, en 1958 se redefinió este concepto (Daniel Koshland) proponiendo que en la fase inicial de la unión la complementariedad es relativamente pobre, pero que tras esa unión inicial la proteína sufre un cambio conformacional que "ajusta" la forma del sitio de unión a la del ligando. Es la hipótesis del "ajuste inducido". Una vez que se ha unido el ligando a su receptor una de las siguientes acciones se lleva a cabo: (1) El ligando no se modifica. Ejemplo transporte (2) El ligando es modificado químicamente. Ejemplo enzimas (3) La proteína sufre un cambio conformacional. Los cambios conformacionales en el ligando están ligados a fenómemos tan importantes como: a) Modificacion de la afinidad de otro sitio de union de ligandos (Alosterismo → cooperatividad) b) Modificacion de la conformacion del sitio catalitico de una enzima (activacion o inhibicion alosterica). Estos los cambios conformacionales inducidos son importantes para (i) - el control de los procesos de control metabolico, (ii) - la regulacion de la expresion genica, (iii) - el control hormonal. Para el caso en que la proteína tenga un único sitio de unión, y si en el medio inicialmente la proteína (P) y el ligando (L) se encuentran libres, al cabo de un tiempo se habrá establecido un equilibrio entre P y L y el complejo proteína-ligando, el equilibrio químico se puede escribir como sigue:
P + L
⇄K1
K-1
PL
Donde K1 es la constante cinética de la asociación cuyas dimensiones son (mole-1 s-1), y K -1 es la constante de velocidad de la reacción de disociación de PL cuya dimension es el inverso de tiempo (s-1). Así: V-1 = V1
K -1[PL] = K1[P][L]
K -1
K1
=
[P][L]
[PL]
= Kd
Kd es la denominada Constante de Disociación y corresponde a la constante de equilibrio de la disociación del complejo PL. No confundir con K -1 que es la constante de velocidad para la reacción de disociación. Kd tiene unidades de concentración. En general [P]total = [PL] + [P]libre luego [P]libre = [P]total - [PL] y por lo tanto:
Kd =
[P]libre[L]
[PL]
Así, el valor de Kd para una proteína y ligando concretos depende críticamente de las condiciones experimentales tales como: pH, fuerza iónica, temperatura o presencia de otros solutos en el medio. Esto es lógico si recordamos que todos los factores anteriormente señalados influyen sobre la estructura 3D de las proteínas, y que el sitio de unión es una zona concreta con una estructura definida dentro de la molécula de proteína. Relación entre Kd y la variación de energía libre en la asociación. Si necesitamos conocer la variación de la energía libre que se produce en la formación del complejo [PL], hay que tomar en cuenta las situaciones inicial y final en el sistema proteína y ligando (sin olvidar las moléculas de agua en la que ambos se encuentran disueltos), dado que el sistema puede estar en dos estados: (I) la proteína y el ligando están separados e interaccionando con las moléculas de agua, y (II) se ha formado el complejo PL y las moléculas de agua que antes estaban unidas a la proteína y al ligando ahora son parte del bulto del disolvente. Es muy importante conciderar que la variación de la energía libre depende, no solo de los enlaces iónicos, de los puentes de hidrógeno y de las interacciones de van der Walls que se formen entre la proteína y el ligando, sino también de las interacciones que tengan con las moléculas de agua y entre las moléculas de agua. La variación de la energía libre en condiciones estándar (concentración de 1 M de reactantes, pH 7,0, 25º C) está relacionada con la constante de equilibrio de la reacción por la expresión: ΔG0' = -RT lnKeq; siendo R la constante de los gases y T la temperatura absoluta. Por lo tanto, si queremos conocer la energía que se libera cuando se asocian la proteína y el ligando, suponiendo que el sistema se encuentra en equilibrio, y teniendo en cuenta que vamos a emplear habitualmente Kd, que es la constante de equilibrio de disociación, tendríamos que emplear la siguiente relación: ΔG0' = -RT lnKasociación =
+RT lnKd;
Para una mejor comprensión de la teoría y aplicaciones del cálculo de la Energía Libre en la Química y la Biología se recomienda revisar el contenido del libro Free Energy Calculations: Theory and Applications in Chemistry and Biology.
|
|
Copyright © Ramón Garduño-Juárez. All Rights Reserved. |
 E-mail: | Mayo 2017
E-mail: | Mayo 2017


 Inicio
Inicio